Lernziel
- Das Programm PyMOL bedienen
- Proteinstrukturen in der PDB-Datenbank finden
- spezielle Aspekte einer Proteinstruktur darstellen und hervorheben
- sicher Proteinstrukturen und Sie interessierende Aspekte darstellen
Um diese Übungen ausführen zu können, müssen Sie sich eine Lizenz besorgen und das Programm installieren. Hier finden Sie Informationen zum Beantragen der Lizenz und zum Download.
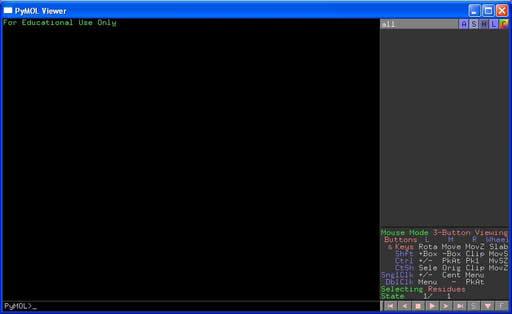
Grafik-Fenster und
Menü-Fenster
Zum Grafik-Fenster
gehören das
Anzeigefenster und die Auswahlliste
Starten Sie nun das Programm. Es erscheint die grafische Benutzeroberfläche, welche aus zwei Fenstern besteht. Der PyMOL Viewer ist das Grafik-Fenster, in dem die 3D-Strukturen dargestellt werden. Es ist wiederum unterteilt in vier Bereiche, wobei uns zunächst nur das Anzeigefenster (links) und die Auswahlliste (oberer Teil recht) interessieren.
Das zweite, kleinere Menü-Fenster,
überschrieben mit The PyMOL Molecular Graphics System,
umfasst die Kommandozeile sowie mehrere Pulldown-Menüs zur
Steuerung von PyMOL.
Strukturdatensätze sind, wie Sie bereits wissen, in
PDB-Dateien zusammengefasst. Ehe Sie mit einem Datensatz arbeiten
können, müssen Sie ihn auf Ihren Rechner kopieren.
Kopieren Sie auf Ihren Rechner den Datensatz für das
TIM-Barrel-Protein Triosephosphat Isomerase von Thermotoga
maritima. Der Datensatz hat den PDB-Code 1b9b. Geben Sie diesen
Code auf der Eingangsseite der Datenbank ein und klicken Sie auf
die Search-Taste.
Studieren Sie zunächst die Angaben, die Ihnen die PDB-Datenbank zu
dieser Struktur liefert.
Wählen Sie dann in der Rubrik Download Files,
die rechts neben dem PDB-Code angegeben ist, die Option PDB-File und speichern Sie die Datei in einem Verzeichnis
Ihrer Wahl.
Falls Sie den PDB-Code der Struktur, die Sie untersuchen wollen, bereits kennen, so können Sie diese auch direkt in PyMOL über Plugin/PDB Loader Service laden. Dabei wird jedoch KEINE Datei auf dem Filesystem angelegt.
Aktivieren Sie nun das Menüfenster von PyMOL und
benutzen Sie File/Open..., um die
soeben deponierte Datei in den Viewer zu laden.

Sie sehen nun die 3D-Struktur der Triosephosphat Isomerase, und zwar
Kette A und B in der sogenannten Linien-Darstellung.
Machen Sie sich nun mit den ersten Befehlen zum Bewegen und
Rotieren der Struktur vertraut. Die Steuerung erfolgt über die
Maus in Kombination mit den drei Maustasten.
Bei gedrückter linker Maustaste können Sie die Struktur um die X-, Y- und Z-Achse rotieren.
Durch Drücken der rechten Maustaste können Sie die Struktur in Z-Richtung (also senkrecht zur Bildschirmebene) bewegen.
Die gedrückte mittlere Maustaste (Mausrad) erlaubt ihnen die Bewegung in der X-Y-Ebene.
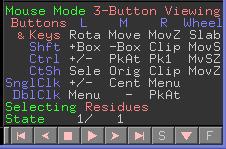
Eine Zusammenfassung der Maus-Befehle finden Sie im PyMOL Viewer rechts unten.
Zusätzlich können Sie über das Main
Pop-Up-Menü (Rechtsklick im schwarzen Hintergrund-Bereich des
PyMol Viewers) /orient (vis) die Struktur automatisch von
PyMOL formatfüllend platzieren lassen.
Betrachten Sie das Protein von allen Seiten, suchen Sie nach Tyrosin-Resten, die in das Lösungsmittel zeigen.
Um die Struktur wieder in die Ausgangsposition zu überführen, wenden Sie bitte den orient-Befehl an.
Diese Befehle sollten Sie in Zukunft immer verwenden, wenn Sie wichtige Details einer Proteinstruktur herausarbeiten wollen.
Um einzelne Residuen, Sekundärstrukturelemente oder Ketten anzusprechen bzw. auszuwählen, bietet PyMOL mehrere Möglichkeiten.
Eine direkte Auswahl von Atomen/Residuen/Ketten geschieht durch Klicken auf ein Atom der entsprechenden Gruppe im Viewer. Auf welcher Auswahlebene Sie sich befinden wird im Mausbefehl-Bereich angezeigt.
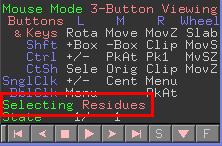
Sie können durch Klicken auf diese Anzeige durch die Auswahlmöglichkeiten blättern (Residues, Chains, Segments, etc.).
Im Moment ist noch kein Residuum ausgewählt.
Zur Zeit werden noch beide Ketten des Proteins angezeigt, die in der PDB-Datei hinterlegt sind. Wir wollen uns im Weiteren auf Kette A beschränken.
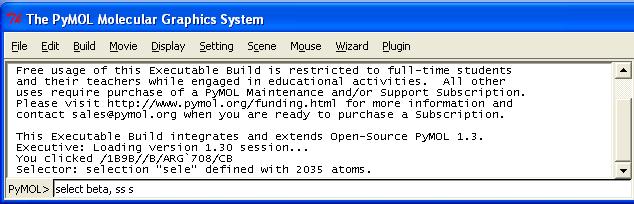
Wählen Sie darum nun die Auswahlebene Kette (Selecting Chains) und selektieren Sie Kette B im Viewer mittels Linksklick auf eines der Atome. Ob Sie die richtige Kette getroffen haben, können Sie im Menü-Fenster kontrollieren. Dort werden alle Auswahloperationen, die Sie mit der Maus tätigen, kommentiert. Die letzten zwei Zeilen der Ausgabe sollten in etwa wie folgt aussehen:
You clicked /1B9B//B/GLY`654/N
Selector: selection "sele" defined with 2035 atoms.
Die Ausgabe beschreibt das Atom, welches Sie zur Auswahl geklickt haben. Der Kettenname steht dabei an zweiter Stelle: /1B9B//B/
Falls Sie Kette A selektiert haben, können Sie die
Auswahl durch Linksklick auf den schwarzen Hintergrund aufheben und einen
weiteren Versuch am anderen Ende der Struktur versuchen.
In jedem Fall taucht in der Auswahlliste oben rechts im Pymol Viewer ein neuer Eintrag namens (sele) auf. Unter Verwendung dieses Eintrags können Sie zahlreiche Operationen auf diese Auswahl anwenden.
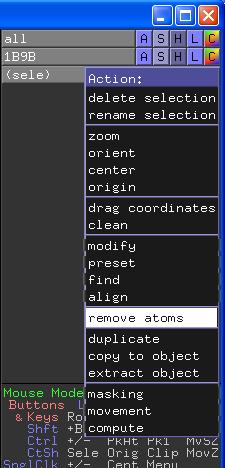
Klicken Sie dazu auf das A (für Action) neben (sele) und wählen Sie im Action-Menü die Option remove atoms.
Einschub:
PyMOL Sessions
In PyMOL steht uns leider keine Undo-Funktion zur Verfügung, so dass manche Aktionen, wie z.B. das Entfernen von Atomen, nicht umkehrbar sind. Stattdessen gibt es aber die Möglichkeit, sogenannte PyMOL Sessions zu speichern und den bisherigen Arbeitsschritte zu sichern bzw. später zu einem gewählten Zeitpunkt zurückzukehren.
Unter File/Save Session As... können Sie den aktuellen Bearbeitungszustand des Datensatzes sichern und diesen später über File/Open... wieder laden.
Dazu nutzen wir die Möglichkeiten der Kommandozeile, um eine Selektion anzulegen. Der allgemeine Befehl lautet:
select Auswahlname, Auswahlkriterien
Um die Beta-Stränge der Kette A anzuwählen müssen Sie als Auswahlkriterium die Eigenschaft Sekundärstruktur angeben:
select beta, ss s
Dabei ist "beta" der Name der Auswahl, den Sie beliebig ersetzen können. Das Argument "ss s" steht für "secondary structure sheet" und selektiert alle Residuen die zu Beta-Strängen gehören. Als weitere Einteilungen stehen h(elix), l(oop) und ""(ohne Zuordnung zu einer Sekundärstruktur) zur Verfügung. Geben Sie nun obigen Befehl auf der PyMol-Kommandozeile ein.
Die Atome, welche zur Auswahl gehören, sind jetzt per Quadrat markiert. Allerdings sind die Sekundärstrukturelemente in dieser Darstellung kaum zu erkennen. Deswegen wechseln wir nun in die sogenannte Cartoon-Darstellung.
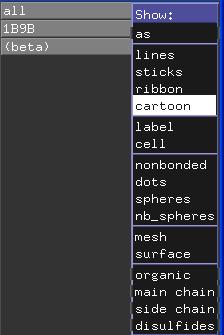
Klicken Sie dazu auf das "S" (für Show) neben "all" in der Auswahlliste und wählen Sie im Show-Menü cartoon.
Um die Übersichtlichkeit noch zu erhöhen, deaktivieren wir noch die Lines-Darstellung. Dazu wählen Sie im H(ide)-Menü bei "all" die Option lines.
Abschließend heben wir die Beta-Stränge noch zusätzlich hervor, indem wir sie einfärben. Da sich die Änderungen nur auf einen Teil der Struktur beziehen sollen, müssen Sie nun auf das bunte "C" (für Color) in der Zeile (beta) klicken und eine Farbe auswählen.
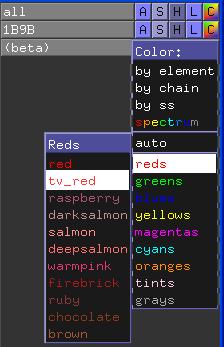
Wir haben nun gelernt, wie Residuen per Kommandozeilen-Befehl und durch Anklicken im Vierwer-Fenster ausgewählt werden können. Zusätzlich können Residuen auch in der Proteinsequenz selektiert werden. Zunächst müssen wir die Sequenz darstellen.
Klicken Sie dazu auf das "S" (für Sequence) in der letzten Zeile rechts unten im Viewer-Fenster.
Über der Proteinstruktur wird nun die Sequenz angezeigt. Die Nummerierung entspricht den Einträgen aus der DB-Datei. Da die erste Aminosäure in der Struktur fehlt, beginnt die Auflistung hier mit 2 bzw. T
Die Residuen sind dabei genau so gefärbt wie in der Struktur, somit sollte es Ihnen leicht fallen, die Positionen zu identifizieren, die dem ersten Beta-Strang entsprechen.
Wenn Sie den Scrollbalken, der zur Sequenzdarstellung gehört, ganz nach rechts schieben, sehen Sie hinter der Sequenz zusätzliche Einträge für ein Sulfat-Ion (SO4) und Wassermoleküle(mit einem O angegeben), die ebenfalls in diesem PDB-Datensatz enthalten sind.
Selektieren Sie nun das Residuum VAL24 und versehen Sie es mit einem Label. Klicken Sie dazu auf das "L"(abel) der Selektion (sele) und wählen Sie die Option residues.
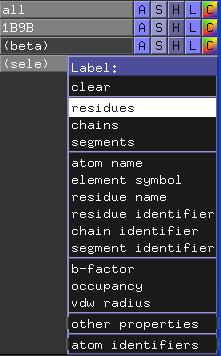
Lassen Sie sich dieses Residuum in der Stick-Darstellung anzeigen.
Um eine größere Menge von Residuen auszuwählen, müssen diese nur angeklickt werden. Sie werden automatisch zur Selektion (sele) hinzugefügt. Allerdings ist die Cartoon-Darstellung dafür ungeeignet, da ein Residuum dann ausgewählt wird, wenn eines seiner Atome getroffen wurde und diese in der Cartoon-Darstellung nicht angezeigt werden.
Stattdessen können Sie auf Residuen-Ebene eine erweiterte Auswahl erstellen, indem Sie in der Sequenz-Zeile mehrere Positionen anwählen. Diese werden, wie oben, automatisch zur Auswahl hinzugefügt.
Um zwei oder mehr Selektionen gleichzeitig erstellen zu können, muss die (sele) Auswahl umbenannt werden, da sie sonst überschrieben wird. Dazu müssen Sie im A(ction)-Menü die Option rename selection ausführen und einen neuen Namen eingeben.
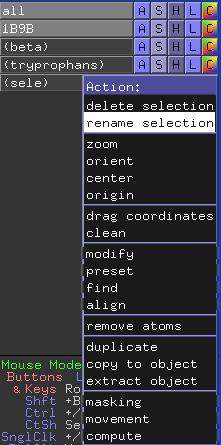
In einer PyMOL-Sitzung können mehrere PDB-Dateien geladen werden, um z. B. Strukturen zu vergleichen oder zu superpositionieren (überlagern). Oft sollen nicht alle Strukturen gleichzeitig dargestellt oder bewegt werden. Daher können in der Auswahlliste einzelne Strukturen an- oder abgewählt und somit die Darstellung kontrolliert werden.
Geben Sie anschließend den Befehl
align 1B9B, 1THF
in die Kommandozeile ein. Sie sehen nun die Überlagerung der beiden Strukturen. Besitzen sie einen gemeinsamen Faltungstyp?
PyMOL eignet sich nicht nur zum Betrachten von 3D-Strukturen, sondern aufgrund der eingebauten Raytracing-Funktionalität auch zum Erstellen von Abbildungen für Vorträge und Veröffentlichungen.
Entfernen Sie zunächst die 1THF-Struktur über A(ction)/delete object. Suchen Sie anschließend eine Ansicht für 1B9B, in der der Aufbau des Proteins aus Sekundärstrukturelementen gut sichtbar ist.
Wenn Sie eine gewünschte Darstellung gefunden haben, so können Sie mittels File/Save Image As/PNG... die aktuelle Ansicht im Viewer speichern.

Die Standard-Darstellung in PyMOL eignet sich allerdings in den seltensten Fällen für qualitativ ansprechende Bilder. Zum einen ist es meist vorteilhaft, die Hintergrundfarbe von schwarz auf weiß zu ändern. Dies erreichen Sie über Display/Background/White oder den Befehl bg_color white in der Kommandozeile.
Wählen Sie nun für eine Struktur die Darstellung spheres und färben Sie sie einheitlich in einer Farbe ihrer Wahl. Speichern Sie die aktuelle Ansicht als PNG-Datei.
Um die Qualität der Abbildung zu erhöhen verwenden wir jetzt das Raytracing-Modul. Klicken Sie dazu auf den Ray-Button im Menüfenster. Nach Ende der Berechnung sehen Sie nun das verbesserte Bild. Klicken Sie nun NICHT mehr in den Bildbereich, da sonst die Ergebnisse verworfen werden und PyMOL zur Standarddarstellung zurückkehrt. Speichern Sie wiederum die Ansicht und vergleichen Sie beide Bilder.
Im Menü Wizard bietet der Befehl Measurement die Möglichkeit Abstände und Winkel in der Struktur zu messen.
Versetzen Sie zunächst PyMOL zurück in den Startzustand mittels File/Reinitialize. Laden Sie anschließend diese PyMOL-Session.
Starten Sie nun die Messmethode unter Wizard/Measurement. Standardmäßig ist die Abstandmessung aktiviert. Klicken Sie auf zwei Atome, deren Abstand Sie bestimmen möchten. Es wird die Verbindungslinie und der Abstand (in Angström) angezeigt.
Falls die Labels nicht sichtbar sind, ändern Sie bitte die Farbe der Labels mit set label_color, yellow.
Zum Beenden des Distance Wizards klicken Sie auf die Done-Taste.

Begründen Sie, weshalb das Protein eine (βα)8-Fass Topologie besitzt.
Das Fluoreszenzsignal von Tryptophan-Residuen ist davon
abhängig, ob sie Wasser (dem Lösungsmittel) ausgesetzt sind, oder im Inneren von
Proteinen liegen. Wir wollen die Lage solcher Reste in diesem Protein
untersuchen.
Verwenden Sie hierfür die Kommandozeile zur Auswahl der Residuen:
select tryptophans, resn trp
Lassen Sie sich die ausgewählten Residuen in der Sticks-Darstellung anzeigen und färben Sie die Tryptophane mit einer auffälligen Farbe ein.
Was schließen Sie für das zu erwartende Fluoreszenzsignal?
Um einen tieferen, aber keineswegs erschöpfenden Einblick in die Funktionalität von PyMOL zu ermöglichen, bietet das Programm einige Demonstrationsfälle, welche Sie im Menü Wizard/Demo finden und ausprobieren können.

Was Sie jetzt verstanden haben sollten