Sowohl für Protein- aber auch RNA-Sequenzen steigt die Vorhersagequalität, wenn als Eingabe ein multiples Sequenzalignment (MSA) anstelle einer einfachen Sequenz verwendet wird. Durch die spaltenspezifischen Häufigkeiten wird für die einzelnen Positionen präziser vorgegeben, welche Ansprüche die Nukleotide bzw. die Aminosäuren erfüllen müssen.
Für eine einzelne RNA-Sequenz wird die 2D-Struktur mithilfe eines Ansatzes der dynamischen Programmierung vorhergesagt. Die Scores sind aus den Bindungsenergien abgeleitet und es werden weitere Regeln, die sich aus realen RNA-Strukturen ergeben, berücksichtigt. Wichtige Implementationen sind der Mfold Server von M. Zuker und die Algorithmen des Vienna-Package. Letzteres enthält eine große Anzahl nützlicher Routinen.
Lernziel
- eine Vorstellung von der 3D-Struktur von RNA haben,
- die vorgestellten Algorithmen zur 2D-Strukturvorhersage von RNA verstanden haben,
- unterschiedliche Darstellungsarten interpretieren können.
| >2D_RNA_1 GCGAAUUUAGCUCAGUUGGGAGAGCGCCAGACUGAAGAUCUGGAGGUCCUGUGUUC GAUCCACAGAAUUCGCACCA |
|
||||||||||||
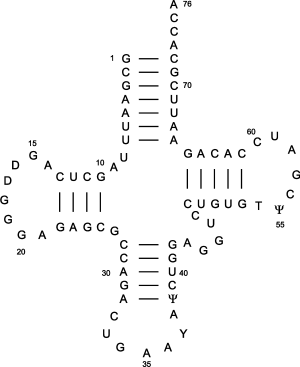
Benutzen Sie den RNAfold-Server des
Vienna-Packages mit
Default-Einstellungen und vergleichen Sie den Graphical output
mit obiger Struktur.
Achten Sie insbesondere auf Helices: Sind diese
korrekt vorhergesagt?
Was bedeutet die Farbe des Hintergrundes der
einzelnen Nukleotide?
MALAT1 ist eine long noncoding RNA von der bekannt ist, dass sie in vielen humanen Krebserkrankungen fehlreguliert ist. Eine kurze, stark konservierte small RNA der Länge 61 Nukleotide, die aus MALAT1 stammt, wird in vielen Geweben exprimiert. Diese RNA wurde mascRNA genannt. Weitere Details werden in dieser Publikation beschrieben.
Hier finden Sie die komplette lncRNA.
Klicken Sie auf diesen Verweis und ändern Sie dann die Darstellung auf FASTA, indem Sie unten rechts im Browser bei Display: auf FASTA klicken.
Bitte achten Sie auf die von den Servern geforderten Dateiformate (reine Sequenz/Fasta-Format).
Vergleichen Sie Ihre Ergebnisse mit den Befunden aus der oben genannten Publikation.
Stimmen die Vorhersagen überein? Wie wurde in der Publikation die 2D-Struktur vorhergesagt?
Falls die Ergebnisse abweichen: Benutzen Sie beim Mfold-Server zusätzlich die Energien der Version 2.3.
Achten Sie bei den RNAfold-Ergebnissen auf die Farbe des Hintergrundes der einzelnen Nukleotide.
Ähnlich wie bei der Vorhersage der Protein-2D-Struktur steigt die Vorhersagequalität, wenn anstelle einer Sequenz ein MSA benutzt wird. Allerdings ist der Performanzgewinn bei den RNA-Sequenzen geringer als bei den Proteinsequenzen:
Wie Benchmarktest gezeit haben, ist die Qualität einer 2D-Vorhersage, die auf einem RNA-MSA basiert, mit der vergleichbar, die für EINE Proteinsequenz zu erwarten ist.
In dieser Übung soll für eine humane ncRNA die Sekundärstruktur zuverlässig vorhergesagt werden. Genauer untersucht wird die RNA U1, die Teil des Spliceosoms ist. Dieser RNA/Proteinkomplex ist, wie der Name vermuten lässt, am Spleißen beteiligt. Dies ist der Prozess, bei dem die Introns aus der prä-mRNA entfernt und die Exons verknüpft werden. Einen Übersichtsartikel zu diesem faszinierenden Komplex finden Sie hier.
Proteinsequenzen werden in PFAM-Datenbank zu Familien zusammengefasst. Analog werden RNA-Sequenzen in der Rfam-Datenbank gesammelt und gruppiert. Darin finden sich z. B. in der Familie RF00003 die korrespondierenden Sequenzen der U1 spliceosomal RNA.
Auf der zugehörigen Seite finden Sie auch die 2D-Struktur der RNA.
Sie sollen an diesem Beispiel den Performanzgewinn untersuchen, den die Analyse eines RNA-MSAs bietet. Die Elemente des Vienna-Packages erlaubt es, eine 2D-Struktur sowohl für eine Sequenz (RNAfold Server) als auch für ein MSA (RNAalifold Server) vorherzusagen. Benutzen Sie beide Algorithmen und vergleichen Sie die Ergebnisse.
Lesen Sie sich zunächst die Beschreibung durch, die Sie auf der Seite zur Familie RF00003 finden, um die Funktion dieser RNA zu verstehen.
Klicken Sie anschließend auf den Reiter Sequences und suchen Sie nach humanen Sequenzen. Sortieren Sie die Sequenzen zunächst nach Type und dann nach Species. Wählen Sie eine seed Sequenz aus Homo sapiens, die möglichst nur die RNA Sequenz enthält. Die Einträge bei Start und End sollten nahe bei 1 bzw. 164 liegen. Sie sollten z. B. diese Sequenz gefunden haben. Speichen Sie die Sequenz lokal ab. Dies ist die eine Eingabe für den Server.
Generieren Sie anschließend ein MSA bestehend aus den "seed sequences" dieser RNA-Familie: Klicken Sie auf der Rfam-Seite auf den Reiter Alignment, ändern Sie das Alignment-Format auf FASTA (gapped), nutzen Sie die Option Download und klicken Sie auf die Taste Generate.
Speichern Sie das Alignment lokal ab. Dies ist die zweite Eingabe für den Server. Ihr Datensatz sollte diesem hier ähneln.
Sie haben nun die beiden Datensätze (einfache Sequenz und MSA) erzeugt.
Lassen Sie vom RNAfold WebServer eine 2D-Struktur für die
einfache Sequenz vorhersagen.
Benutzen Sie die Default-Parameter und
klicken Sie auf die Proceed-Taste.
Notieren Sie zunächst die freie Energie des Ensembles.
Betrachten Sie in der Ausgabe die MFE secondary structure und sichern Sie die PDF Datei, die "MFE structure drawing encoding base-pair probabilities" enthält. Machen Sie sich bitte klar, welche Eigenschaft der Farbcode bewertet.
Lassen Sie sich nun vom Server RNAalifold eine 2D-Struktur für das MSA erzeugen.
Benutzen Sie wiederum die Default-Parameter und klicken Sie auf Proceed.
Notieren Sie wiederum die freie Energie des Ensembles.
Sichern Sie die PDF Datei, die "structure drawing encoding base-pair probabilities" enthält.
und bewerten
Welches Ensemble besitzt die "bessere" Energie?
Hat sich die Vorhersagequalität (Energie) bei der Verwendung des MSAs verbessert? Vergleichen Sie auch die Werte der base-pair probabilities.
Vergleichen Sie die zwei vorhergesagten 2D-Strukturen mit der, die Sie auf der Rfam-Seite unter bpcons finden.
Welche der beiden Vorhersagen stimmt besser mit dieser Struktur überein? Wo gibt es Unterschiede?
Welche Schlüsse ziehen Sie aus diesem Vergleich für zukünftige Analysen?
Was Sie jetzt verstanden haben sollten