Lernziel
Nach dem Bearbeiten dieser Übungen sollten Sie
- sicher Proteinstrukturen und Sie interessierende Aspekte darstellen,
- ein Basiswissen zu Mutationsanalysen besitzen und die hierbei verwendeten Algorithmen benennen
Zudem sollten Sie die Übungen zur Visualisierung von Protein-3D-Strukturen bereits absolviert haben. Die Installation von PyMol ist hier beschrieben.
Vor Position 120 dieses Enzyms ist bekannt, dass sie bei der Katalyse von zentraler Bedeutung ist. Zudem ist die Besetzung dieser Position strikt konserviert: In einem multiplen Sequenzalignment von Metallo-β-Lactamasen findet sich dort nur Asp.
Die genaue Rolle dieses Residuums bei der Enzymfunktion war lange Zeit unklar. Üblicherweise werden in solchen Fällen Protein-Mutanten hergestellt und vermessen. Im konkreten Fall werden an Position 120 mithilfe molekularbiologischer Methoden unterschiedliche Reste eingeführt und die Veränderungen in der Enzymfunktion mit biochemischen und biophysikalischen Methoden studiert.
In dieser Übung betrachten wir zunächst das aktive Zentrum des Enzyms. Wir gehen hierbei von einer Mutante aus, in der das Asp an Position 120 durch Serin ersetzt wurde. Diese Arbeiten sind in dieser Publikation beschrieben, an der wir uns im Folgenden orientieren. Als Ergebnis der genannten Studien wurde postuliert, dass Asp-120 als starker Zink-Ligand wirkt, das Ion optimal im Hinblick auf Substratbindung positioniert und an der Ausbildung einer partiellen negativen Ladung beteiligt ist.
Aktivieren Sie dazu die Sequenzanzeige. Der Datensatz enthält eine Kette (A) sowie zwei Hetero-Atome (Zink), die zum Reaktionszentrum gehören. Diese Atome werden ganz rechts in der Liste aufgeführt.
Klicken Sie dazu in der Auswahlliste auf das S neben 2UYXmod und wählen Sie as/cartoon.
Verändern Sie die Farbe der Cartoon-Darstellung z.B. mit C/by ss/Helix Sheet Loop.
Selektieren Sie beide Zn-Atome in der Sequenzzeile. Sie werden nun als Selektion (sele) in der Auswahlliste angezeigt. Wählen Sie für diese Auswahl S/as/spheres und kolorieren Sie die beiden Atome in der Farbe gray50.
Außerdem liegt Arg121 in unmittelbarer
Nähe der Zink-Atome.
Weiterhin sind zwei Wassermoleküle in die Bindung
mittels Wasserstoffbrückenbindungen involviert, diese
Wassermoleküle sind in dieser Kristallstruktur allerdings nicht
sichtbar.
Bindungen können nur ausgebildet werden, wenn bestimmte
Abstandskriterien erfüllt werden. Wir wollen daher alle Residuen
bestimmen, die maximal 4 Å von Zinkatomen entfernt liegen.
Stellen Sie sicher, dass die Selektion (sele)
nur die beiden Zink-Atome umfasst.
Benutzen Sie anschließend für (sele) die Option A/modify/around/residues within 4 A.
Aufgrund der
Mutation wird die Position 120 nicht gefunden. Reinitialisieren Sie zunächst PyMOL und laden Sie danach
den Datensatz 2UYXmod.pdb neu. Lassen Sie sämtliche Wasserstoffatome, die im
PDB-Datensatz nicht enthalten sind, hinzufügen mit A/hydrogens/add.
Die Existenz dieser Wasserstoffe ist für die folgenden Analysen
wichtig. Deaktivieren Sie nun jegliche Darstellung (H/everything). Wir wollen nun auch die unmittelbare 3D-Umgebung des Residuums
darstellen lassen. Selektieren Sie dazu alle Residuen im Umkreis von 6 Angström um Ser120. Aktivieren Sie für
diese Selektion die Lines-Darstellung
und ändern Sie die Farbe zu grau. Lassen Sie von den Zinkatomen die van der Waals
Oberflächen in braun darstellen (zu finden in der Farbgruppe rot). Benutzen Sie die Funktion Wizard/Mutagenesis
und picken Sie die Ser120-Seitenkette. Ändern Sie die
Aminosäure von No Mutation zu Mutate
to ASP. Die für jedes Rotamer zusätzlich angezeigten
farbigen Scheiben und Striche bedeuten folgendes: Entscheiden Sie sich für ein Rotamer, dass
möglichst wenig sterische Probleme einführt. Bestätigen Sie ihre Wahl mit Apply.
Erst dann wird die Mutation wirklich durchgeführt. Mit Done
können Sie den Mutagenesis Wizard beenden. Sie können Ihre Rotamerwahl noch zusätzlich auf
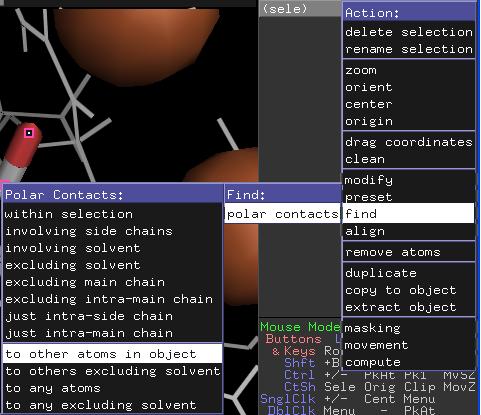
energetisch günstige polare Interaktionen überprüfen. Selektieren Sie dazu das mutierte Residuum Asp120 und
wählen Sie die Option A/find/polar
contacts/to other atoms in object. Es sollten nun
mindestens drei, optimalerweise vier Wechselwirkungen angezeigt werden. Falls nicht,
überprüfen Sie nochmals die Rotamerwahl. Im Viewer sind nun zwei Datensätze geladen. Zunächst
wollen wir die Datensätze superpositionieren und uns dabei auf die
CA-Atome beschränken. Der Kommandozeilenbefehl dafür lautet: align (2B2C and name ca), (2UYXmod and name ca) In der Anzeige des Menüfensters wird mit dem RMSD-Wert
ein Maß für die Qualität der Überlagerung
angegeben. Der Wert von 0.257 Angström entspricht der mittleren
Abweichung von 185 einander zugeordneten CA-Atomen und somit einer fast
perfekten Überlagerung. Die bisher erzielten Ergebnisse belegen, dass wir
in der Lage sind, mit relativ einfachen Mitteln die Position der
Seitenketten zumindest annähernd zu bestimmen. Allerdings
können wir anhand dieser Betrachtung keine Aussage über den
Effekt der Mutation auf die Stabilität des Proteins oder die
Enzymaktivität machen. Für eine genauere Bewertung des Einflusses von Mutationen
auf die Proteinstabilitäten bietet die Bioinformatik andere
Werkzeuge. Zu den bekanntesten Programmpaketen für die Zwecke des
Enzymdesigns gehört RosettaDesign,
das auch als frei verfügbarer Webserver genutzt werden kann. Sie
können sich dort anmelden und eine PDB-Datei zusammen mit einer
Liste der gewünschten Mutationen übergeben. Je nach Auslastung müssen Sie auf die Ergebnisse mehrere
Tage warten. Daher finden Sie hier bereits die Ergebnisse für die Ausgangsstruktur (2UYX) und die Punktmutante (S120D), die dem wildtypischem
Enzym entspricht. Speichern Sie die beiden Dateien lokal und
öffnen Sie diese mit einem Texteditor (z.B. Notepad++). Die Dateien beinhalten zum einen die Struktur im PDB-Format
und im zweiten Abschnitt eine detaillierte Tabelle mit zahlreichen
Energiewerten. Die für Sie relevanten Informationen finden Sie in
den Zeilen beginnend mit pose und SER_81
bzw. ASP_81. Der Wert für die
Gesamtenergie steht jeweils am Ende der Zeile. Die Energiewerte (Rosetta-Scores) können NICHT direkt
physikalisch interpretiert werden, allerdings gilt auch hier, dass eine
negativerer Wert einer höheren Proteinstabilität entspricht. Welchen Effekt auf die Stabilität des Proteins
erwarten Sie durch die Punktmutation? Beachten Sie, dass die Aminosäure Asp an
dieser Stelle in natürlich vorkommenden Enzymen streng konserviert
ist. Um die Übereinstimmung der aktiven Zentren zu
überprüfen, sollen Sie sich folgende Residuen und
Hetero-Atome anzeigen lassen: Färben Sie beide aktive Zentren unterschiedlich ein. Was Sie jetzt verstanden haben sollten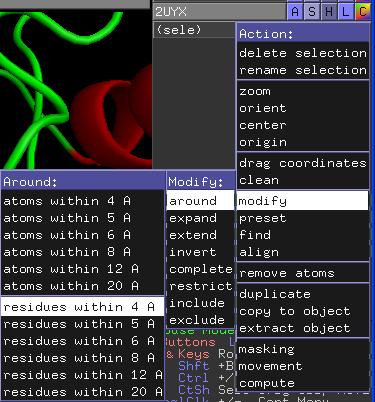
Überprüfen Sie nun in der
Sequenzzeile, welche Residuen ausgewählt wurden. Lassen Sie sich diese Residuen der Stick-Darstellung
anzeigen.
Sie können sich zusätzlich
als Label die Residuennamen anzeigen lassen (L/residues).
Schalten Sie für das gesamte Protein
die Cartoon-Darstellung ab.
Sie sehen jetzt das aktive Zentrum dieses Enzyms.
Die
dargestellten Residuen sollten mit denen übereinstimmen, die oben
genannt wurden. Vergleichen Sie hierfür die
Label mit obiger Liste.
Lassen Sie
zusätzlich Haupt- und Seitenkette von Ser120 darstellen und markieren Sie
diese in blau.
Hier finden Sie zur Überprüfung Ihrer Arbeit eine Lösung.
Übung
ProtMut_2,
Mutation einführen und optimieren
Motivation
Die Methoden der
Mutationsanalyse, der Energieminimierung und ganz allgemein Molecular
Modelling hat das Ziel, die 3D-Struktur von Proteinen unter
Verwendung von Kraftfeldern präzise vorherzusagen.
In dieser Übung wollen wir Möglichkeiten und Grenzen dieser
Algorithmen untersuchen. Das von uns verwendete Verfahren ist ein
einfaches, da für eine fundierte Analyse ausführliche
Simulationen mit entsprechenden Laufzeiten auf größeren
Rechnern erforderlich sind. Daher verwenden wir hier Funktionen, die in PyMOL implementiert sind.
Das Protein, das wir
oben untersucht haben, enthält eine Punktmutation an Position 120.
Das wildtypische Enzym aus Bacillus cereus besitzt an dieser
Stelle ein Asp.
Da die Struktur dieses wildtypischen Enzyms bekannt ist, wollen wir in
obiger Mutante diese Veränderung rückgängig machen und
anschließend untersuchen, ob die Orientierung der
Seitenkettenatome, die sich nach Energieminimierung ergeben, mit der im
Wildtyp übereinstimmt.
Positionieren
des Proteins
Lassen Sie Residuum Ser120 in Stick-Darstellung
und in rot anzeigen.
Verändern Sie anschließend die Darstellung so, dass das
rot-markierte Ser120 mittig und gut sichtbar im Darstellungsfenster
liegt. Zoomen Sie soweit in das Protein, dass Sie nur noch die
nähere Umgebung dieses Residuum sehen und positionieren Sie das
Protein so, dass die Ser-Seitenkette möglichst frei vor dem
Hintergrund zu liegen kommt.
Mit diesen Ma&szling;nahmen wird das Picken des Restes für die
nachfolgenden Aktionen erleichtert.
Mutation
ausführen
Mutieren Sie die Position 120 zu einem
Asp.
Anleitung
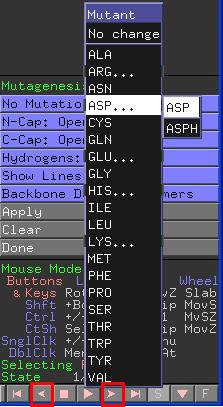
Klicken Sie auf die markierten Pfeile unten rechts im
Viewer-Fenster und beobachten Sie die Darstellung. Es wird jeweils ein
anderes Rotamer der Seitenkette ausgewählt und
dargestellt. Beim Klicken auf den dazwischenliegenden Pfeil wird
automatisch durch alle Lösungen iteriert.
grün: Atome sind fast in Kontakt oder
überlappen sich leicht.
rot: Dieses Rotamer bedingt große
sterische Probleme, die korrigiert werden müssen. Es entstand ein
signifikanter Überlapp der van der Waals Radien.
orange: Eine Situation, die zwischen "rot" und
"grün" liegt.
Polare
Interaktionen
Vergleich mit
der Situation im Wildtyp
Wir wollen nun die
resultierende Lage der Seitenkette mit der im Wildtyp vergleichen.
Laden Sie hierzu den Datensatz 2BC2.pdb, welcher zwei Ketten A und B
umfasst. Die Nummerierung der Reste in diesem Datensatz unterscheidet
sich von der in 2UYX.
Strukturen
optimal überlagern
Allerdings wird die Darstellung im Viewer nun sehr unübersichtlich
sein. Dies müssen wir ändern, da wir nur an der Lage weniger
Seitenketten interessiert sind.
Relevante
Positionen auswählen
Stellen Sie
zunächst fest, mit welcher Kette von 2BC2 PyMOL die 2UYXmod
Struktur
überlagert hat. Deaktivieren Sie anschließend für 2BC2
jegliche Darstellung.
Lassen Sie exklusiv die Seitenkette des Restes Asp90
(richtige Kette wählen!) in der Stick-Darstellung anzeigen und
markieren Sie ihn grün. Diese Position entspricht der Position 120
in der Mutante.
Vergleichen Sie die Ausrichtung der
Seitenkette in beiden Strukturen.
Ergebnis
Sie erkennen, dass die
oben gewählte Rotamerausrichtung gut passt, aber nicht exakt der
des Wildtyps entspricht.
Dies kann mehrere Gründe haben. Die
wichtigsten sind:
1) Die beiden Wasser-Moleküle, die für die Positionierung der
Zink-Atome wichtig sind, werden hier nicht berücksichtigt.
2) PyMOL lässt bei der Mutation nur eine kleine Menge häufig
beobachteter, diskreter Rotamere zu, welche nicht notwendigerweise den
lokalen Bedingungen im Protein genügen.
RosettaDesign-Server
Wie passt dieser Befund mit dem vorhergesagten Effekt auf die
Proteinstabilität zusammen?
Aktive Zentren
vergleichen
2UYX: His116, His118, Asp120, His196 und Cys 221, His263, ZN1296,
ZN1297
2BC2: His86, His88, Asp90, His149, CSD168 und His210
Vergleichen Sie die
aktiven Zentren, d. h., die Ausrichtung der Seitenketten. Wo gibt
es Übereinstimmungen, wo treten Unterschiede auf?
Mutationen in Proteinen können mit Hilfe bioinformatischer
Methoden
bewertet werden. Algorithmen zur Berechnung der Energie von Proteinkonformationen werden in der Bioinformatik breit eingesetzt.